Comparative transcriptomics method to infer gene coexpression networks and its applications to maize and rice leaf transcriptomes
比较转录组学方法推断基因共表达网络及其在玉米和水稻叶片转录组中的应用
TO-GCN时序分析,媲美加权基因共表达网络分析-WGCNA分析的另一种方法,有有趣的有意思的挖掘方法文献欢迎评论留言~
重要性
时间序列转录组用于分析生物过程非常有用,可用于识别该过程的调控因子。为分析此类数据,我们开发了一种避免在两种条件下进行时间点对齐和标准化的方法。我们将其应用于分析在光暗循环及全黑暗条件下玉米叶片发育的时间序列转录组,获得了一个时间有序的基因共表达网络(TO-GCN),该网络可能包含了参与克兰茨解剖结构发育的所有基因,克兰茨解剖结构是C4光合作用高效率的关键结构。利用这个TO-GCN,我们预测并实验验证了克兰茨解剖发育的上游调控级联。此外,我们还将该方法应用于比较玉米和水稻叶片段的转录组,并识别了玉米C4酶基因和RUBISCO小亚基2的调节因子,展示了其广泛的实用性。
摘要
在不同条件下获得的生物过程的时间序列转录组有助于识别该过程的调节因子及其调节网络。然而,这类数据是3D的(基因表达、时间和条件),目前还没有方法能够处理它们的全部复杂性。在这里,我们开发了一种避免在条件间进行时间点对齐和标准化的方法。我们将其应用于分析在光暗周期及全黑暗条件下玉米叶片发育的时间序列转录组,获得了八个时间有序的基因共表达网络(TO-GCNs),这些网络可以用来预测GCNs中任何基因的上游调节因子。八个TO-GCNs中的一个是光独立的,可能包括了参与克兰茨解剖结构发展的所有基因,这是C4植物光合作用高效率的关键结构。使用这个TO-GCN,我们预测并实验验证了短根1(SHORTROOT1,克兰茨解剖结构的关键调节因子)上游的调控级联。此外,我们还将该方法应用于比较玉米和水稻叶片段的转录组,识别了玉米C4酶基因和RUBISCO小亚基2的调节因子。我们的研究不仅提供了一种强大的方法,还提供了关于克兰茨解剖结构发展和C4光合作用调节网络的新见解。
在不同条件下获得的同一组织的转录组可以揭示出基于条件特异性反应的差异表达基因。此外,时间序列实验的转录组可以提供信息,帮助了解随时间变化的发育过程的动态调控(1)。这种3D(基因表达、条件和时间)数据对于研究基因调控网络以及生物过程的动态非常有用。注意,“条件”可以替换为“物种”或“菌株”,“时间序列”可以替换为“组织”或“来源”。虽然已经开发了分析3D数据的方法(2, 3),但这些方法并没有处理时间序列数据的全部复杂性。例如,一种方法是将实验中一个基因的时间序列表达水平替换为代表性值,如平均值或最大值(3)。这种方法丢失了时间信息。另一种方法是将多个时间序列数据集融合成一个时间序列(2)。这种方法简化了分析,但产生了在原始数据中不存在的基因表达模式聚类。随着测序技术的进步,转录组数据的呈指数增长,迫切需要一种能够从3D数据中提取有价值信息的方法。
在这项研究中,我们开发了一种比较性的、时间有序的基因共表达网络(TO-GCN)方法来分析3D数据。为了说明其构建和应用,我们使用了两组时间序列转录组,这些转录组来自发育中的玉米叶,从0小时(T00,干种子)到吸水后72小时(T72),一组在自然光暗(LD)循环下(4),另一组在全黑暗(TD;本研究中获得)下。因为玉米在TD下的基因表达动态与在LD下大不相同,直接比较两种条件下的基因表达谱是困难的。我们的方法克服了这一挑战,其应用于上述两个时间序列数据集得到了八个TO-GCN,其中一个是光独立的。由于克兰茨解剖在LD和TD下都在发展(附录SI,图S1),光独立的TO-GCN可能包括了所有参与克兰茨解剖发展的基因,这对于C4光合作用的高效率至关重要。使用这个TO-GCN,我们推断并实验验证了一个关键的克兰茨解剖调节器,即玉米SHORTROOT1(ZmSHR1, Zm00001d021973)的上游调控级联(5)。最后,为了展示我们方法的广泛实用性,我们用它比较了发育中的玉米和水稻叶片的不同部分的转录组(6),并鉴定了编码关键玉米C4酶和RUBISCO小亚基2(ZmRBCS2, Zm00001d004894)的基因的调节因子。因此,我们的研究不仅提供了一种强大的方法,还提供了关于克兰茨解剖发展和C4光合作用调控网络的新见解。
结果
在LD或TD条件下的早期玉米叶片发育
为了说明比较时间序列转录组数据需要新方法的必要性,我们选择研究在自然光暗周期(13小时光照/11小时黑暗;日光从约早上6点到晚上7点)和全黑暗(附录SI,图S1A)条件下玉米幼苗的发育进程。在LD条件下,幼苗经历光形态生成,其特点是具有短的胚根、胚芽鞘、叶片和胚轴,并在吸水后66到72小时胚芽鞘变绿。相比之下,在TD条件下,幼苗经历暗形态生成,其特点是苍白和伸长的胚芽鞘、叶片、胚轴和胚根。众所周知,无论在LD还是TD条件下生长的玉米幼苗都会发展出带有克兰茨标志的维管组织(附录SI,图S1B),因此,比较LD和TD数据有助于排除不参与克兰茨解剖结构发展的大量光调控基因。
玉米叶片转录组
刘等人(4)在LD条件下获得了从干种子(0小时)到吸水后72小时的13个时间点的发育中的胚胎叶片转录组,每6小时采集一次(附录SI,图S1A)。在本研究中,我们在TD条件下从T06到T72获得了一组平行的12个时间点的发育中的胚胎叶片转录组(7)(附录SI,表S1)。通过共享T00(干种子)的转录组,我们拥有在LD和TD条件下的两组13个转录组。序列读取的处理和映射以及RPKM(每百万映射读取的转录本每千碱基的读取量)的标准化在方法部分有描述。我们定义一个基因为表达的,如果它在25个转录组中至少2个的RPKM >1。总共有25,489个基因,包括1,718个转录因子(TF)基因,被认为是表达的,并用于后续分析。
分析3D转录组数据的方法学考虑
胚胎叶片在TD条件下的初期发育速度比在LD条件下快(附录SI,图S1A),因此基因在TD条件下的上调或下调倾向比在LD条件下更早(见例子在附录SI,图S2)。例如,ZmSCR1(SCARECROW;Zm00001d052380),一个在玉米克兰茨叶片解剖发展中的关键TF,在LD条件下在T54被上调,而在TD条件下在T48被上调。一个基因在TD条件下的表达谱可能显示时间位移,即使它与在LD条件下的类似(附录SI,图S2)。因此,在比较两种不同发育程序获得的两组转录组时考虑时间位移效应是很重要的。然而,这并不简单,因为时间位移的程度因基因而异(附录SI,图S2)。为了克服这一点,我们的方法首先考虑在每组时间序列转录组中基因的共表达,然后比较LD和TD条件下的共表达模式(图1A)。具体来说,我们考虑在LD条件下共表达的两个基因是否也在TD条件下共表达,反之亦然。然后我们使用基因共表达关系来构建GCN。此外,由于我们的数据是时间序列数据,我们的方法还可以确定GCN中节点的时间顺序(图1B)。一个时间有序的GCN可以揭示基因功能的动态和生物过程的时间转变(图1B)。下面,我们解释如何计算基因共表达关系,然后构建TO-GCN。
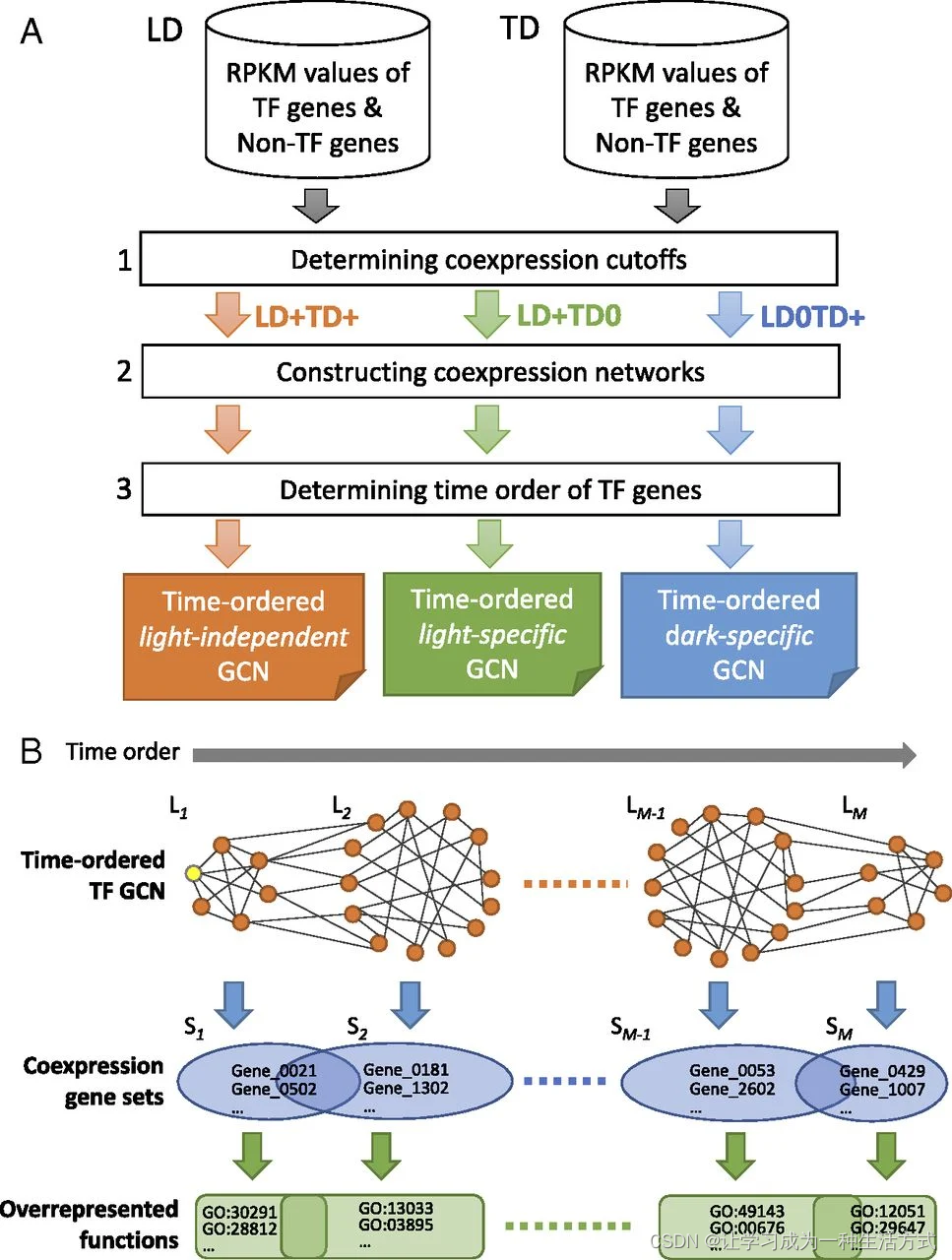
流程图说明比较转录组学方法。(A)构建转录因子时间有序基因共表达网络的三个步骤。展示了光独立(LD+TD+)、特定黑暗(LD0TD+)和特定光照(LD+TD0)的GCN作为三个示例。+/− 表示正/负共表达。(B)TO-GCN中的级别(L1至LM)代表转录因子基因的上调时间顺序,不同级别对应的共表达基因集(S1至SM)(包括非转录因子基因),以及过表达的功能。L1中的黄色节点代表初始节点。一个集合中的基因可能与多个级别的转录因子共表达,因此它们可能属于多个集合。
计算基因共表达关系和网络
为简化起见,我们首先关注转录因子基因。我们定义两个转录因子基因之间的共表达关系,或一个转录因子和一个非转录因子基因之间的共表达关系如下。首先,我们分别在LD和TD条件下计算所有1,718个表达的转录因子基因对的原始RPKM值的皮尔森相关系数(PCC),发现任何两个转录因子基因之间的PCC超过0.84的概率为P<0.05(附录SI,图S3)。对于转录因子和非转录因子基因之间的共表达,阈值相似(即,PCC≥0.83时P<0.05)。因此,我们定义两个基因为正相关共表达(根据数据集分别标记为LD+或TD+),如果PCC≥0.84。我们还定义两个基因为不共表达(标记为LD0或TD0),如果−0.5≤ PCC <0.5,并且定义为负相关共表达(标记为LD−或TD−),如果PCC<−0.75。综合考虑LD和TD下的共表达状态,如果两个基因在LD和TD下均为正相关共表达,则我们称这两个基因属于LD+TD+关系集。类似地,如果两个基因只在LD(或TD)下正相关共表达,而在TD(或LD)下则不是,我们称这两个基因属于LD+TD0(或LD0TD+)。附录SI,表S2显示了每组基因共表达关系中的基因总数和转录因子-基因对(这里的基因可以是转录因子或非转录因子);不共表达基因集(LD0TD0)不在我们的关注范围内。下面,我们只检查LD+TD+,这很可能包括了参与克兰茨解剖结构发展的所有关键基因。其他集将在后续研究中讨论。
在LD+TD+集中,共表达关系独立于光效应,使我们能够缩小克兰茨解剖的候选调节因子。在LD+TD+中的1,275个转录因子基因中,1,207个形成了一个大的主要转录因子GCN,其节点由共表达关系连接。这个GCN被称为光独立的转录因子GCN,并在附录SI,图S4中可视化。其余68个转录因子基因形成28个GCN,每个包含少于10个转录因子基因。由于GCN中连接的转录因子基因在时间序列中具有相似的上调或下调时间点,我们可以推断主要GCN中所有转录因子基因在叶片发育过程中的表达时间顺序。为此,我们选择ZmARF2-1(生长素反应因子2-1;Zm00001d041056)作为初始节点,因为它在LD条件下T00的第一个共表达模块中表达高峰(4),即其表达水平在时间0非常高(RPKM 96)并单调递减至T72。此外,ZmARF2-1是一个生长素反应因子,而生长素是细胞分裂和幼苗发育中的重要植物激素。这些观察结果与我们的假设相符,即ZmARF2-1在萌发过程中发挥作用。然后我们应用广度优先搜索(BFS)算法(9)为主要GCN中的所有转录因子基因分配时间顺序级别(方法)。我们将这个主要GCN称为光独立的转录因子TO-GCN,它由15个时间顺序级别组成(在图2A中标记为L1至L15)。

光独立的TF TO-GCN及不同级别TF基因的标准化基因表达谱。(A) TO-GCN结构,以TF基因为节点(蓝色虚线圆圈)。圆圈中的数字表示该级别上的TF基因数量。(B 和 C) 分别在LD和TD条件下,TO-GCN每个级别的TF基因在每个时间点的平均标准化RPKM(z分数)的热图。对于每个TF基因,首先将时间点上的RPKM标准化为z分数。对于每个级别,热图中所有TF的z分数进行平均。(D) 每个级别共表达基因的过表达MapMan功能。图中的数字(顶部)对应于表中列出的过表达功能的索引号(底部)。蓝色、橙色和绿色分别代表萌发、叶片发育和光合作用阶段。(E) 每个TO-GCN级别上四个激素信号转导途径中基因的比例(数据集S2)。
光独立的TF TO-GCN
如前所述,光独立的TF TO-GCN中的TF基因被分配到15个级别(图2A)。这些分配的级别与LD和TD下13个时间点上TF基因的表达时间顺序相匹配,如每个热图中沿对角线的红色方块(高表达水平)所示(图2B和C)。此外,高表达时间段在连续级别之间重叠,表明某一级别的TF基因可能是下一级别TF基因的调节因子。此外,在TD条件下与LD相比,相同级别的大多数TF基因更早地被上调或下调(图2B和C)。例如,L1级别的TF基因在LD下的T12被下调,而在TD下的T06被下调(图2B和C)。这一观察结果与TD下更快的发育速度一致(附录SI,图S1A)。
在光独立的TF TO-GCN中,许多与种子萌发或打破休眠相关的TF出现在较早的级别(图2A和数据集S1)。例如,ZmABI3(脱落酸不敏感3;Zm00001d042396)和ZmABI5(Zm00001d012296),在胚胎发生期间发挥关键作用(10, 11),属于L2级别。ZmGAI(赤霉素不敏感蛋白;Zm00001d013465),促进种子萌发(12),属于L3级别。这些观察表明我们的方法可以有效揭示种子萌发的关键调节因子。另一方面,所有已知的维管组织发育的正向调节因子都位于较晚的级别,包括两个ZmMP基因(单生叶;Zm00001d001945和Zm00001d026540)和ZmHB8(同源盒基因8;Zm00001d008869)(13),分别属于L12和L11级别。此外,与束鞘(BS)发育相关的正向调节因子ZmSHR基因(5)(Zm00001d021973,Zm00001d029607和Zm00001d006721)和ZmSCR1(8)分别属于L11和L12级别。根据热图中L11的TF基因表达谱(图2B和C),维管组织和外围细胞的发育可能分别在LD和TD条件下的T30和T24开始。因此,与维管组织和BS细胞的发育密切相关的克兰茨解剖结构的关键调节因子应该属于L8、L9或L10。
光独立功能
通过识别光独立TO-GCN中每个级别共表达基因的过表达功能类别(图2A和数据集S2),我们发现L8和L9之间存在明显的发育阶段转变(图2D)。在前八个级别中,蛋白质通过泛素化介导的降解过表达。在前四个级别中,还过表达了其他九个功能,例如蛋白质的翻译后修饰、脱落酸(ABA)的激素代谢、生物和非生物压力、线粒体膜上的代谢物转运蛋白以及钾的运输(图2D),其中一些与种子萌发有关(14)。通过将前八个级别映射到热图中的时间点(图2B和C),我们发现萌发阶段分别在LD和TD下的T48和T30结束。与L1至L8相比,L9至L15的基因主要在与细胞增殖相关的过程中发挥作用,表明在萌发后发生向叶片和根的发展的转变。过表达的功能类别包括在L9和L10的核苷酸合成,在L10至L12的细胞骨架组织,在L11至L13的DNA合成,在L12至L13的细胞周期(图2D)。除了细胞增殖外,与叶绿体发展和光合作用相关的功能在最后四个级别出现。这一观察表明,即使在完全黑暗中,玉米胚胎叶细胞也被编程为发展成能够在光照下进行光合作用的细胞。
植物激素对于触发发育重编程至关重要。我们发现与ABA信号传导相关的基因倾向于在前三个级别出现,但随后减少,这与ABA在萌发开始之前维持种子休眠的作用一致(图2E)。与ABA相反,大多数与赤霉素(GA)信号传导相关的基因出现在L2和L3,这与GA对ABA诱导萌发过程的拮抗作用一致。大多数与生长素和细胞分裂素(CK)信号传导相关的基因出现在L9至L15和L10至L13(图2E)。这也与以往的研究一致,这两种激素共同在调节叶细胞的增殖和分化中发挥主要作用(15)。
Kranz 解剖学的关键调节因子的上游调节因子
已显示ZmSHR1在玉米Kranz 解剖结构的BS细胞发展中起重要作用(8, 16, 17),但其上游调节因子仍然未知。因此,我们选择ZmSHR1作为一个例子,展示我们的方法如何用于识别特定基因的上游调节因子。具体来说,我们使用光独立的TF TO-GCN设计了一个三步工作流程(图3),以识别调节ZmSHR1表达的上游调节路径。
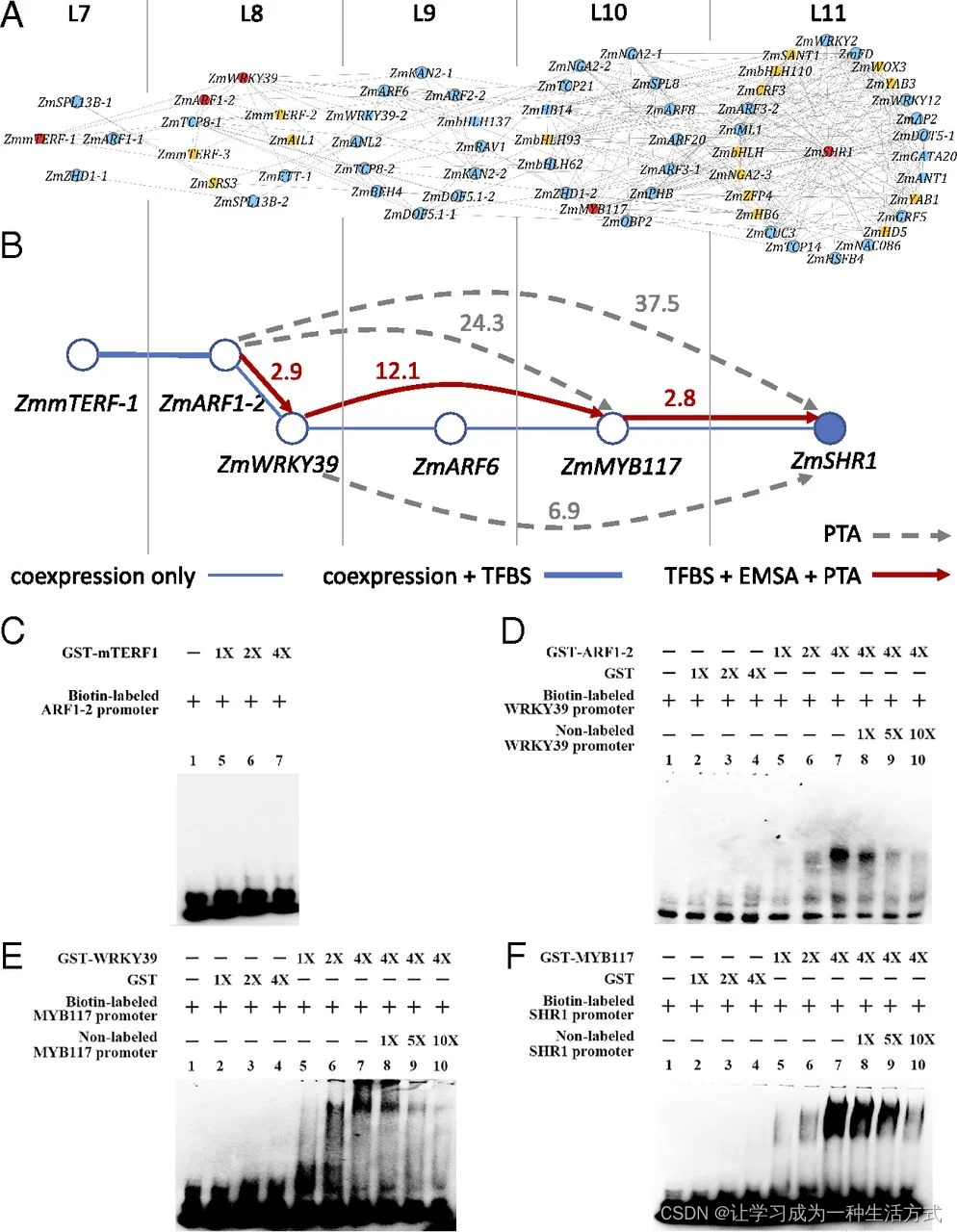
推断和实验验证玉米叶片发育中ZmSHR1基因上游候选调节因子。(A)从光独立的TF TO-GCN推断出的ZmSHR1的第一到第四级候选上游调节因子(附录SI,表S3)。ZmSHR1位于该子网络第11级的中心。未知转录因子结合位点(TFBS)的转录因子以橙色显示。已知TFBS的转录因子(蓝色)用于检查它们在候选下游基因启动子序列中的映射位点。预测的调节途径中具有TFBS支持的转录因子以红色显示,并选定用于实验验证。(B)针对实验验证的ZmSHR1上游调节网络。有向实线或带箭头的虚线旁边的数字表示在PTA实验中潜在直接或间接靶基因表达水平的倍增。红线:靶基因启动子中存在TFBS(由箭头指示),并通过PTA和EMSA验证。灰色虚线:有PTA支持但在启动子中未发现TFBS。(C-F)EMSA实验测试预测的TFBS与推定靶标的启动子中的相互作用以及GST-ZmmTERF1(C),GST-ZmARF1-2(D),GST-ZmWRKY39(E)或GST-ZmMYB117(F)。每种情况下:第1道:生物素标记的探针单独存在;第2到第4道:增加纯化的GST的量(除C外);第5到第7道:增加GST-TF的量;第8到第10道:增加未标记探针的量(除C外)。
首先,我们使用TO-GCN预测ZmSHR1的直接候选调节因子,这些因子应该与ZmSHR1在同一级别或ZmSHR1之前的一个级别共表达(即图3A中的L11和L10)。(一个转录因子可以是同一级别的另一个转录因子的上游调节因子,因为每6小时只取一个转录组。)我们称这些转录因子为一级候选调节因子。类似地,我们推断L9、L8和L7的二级、三级和四级候选调节因子(图3A)。这些候选调节因子列在附录SI,表S3中。
其次,对于每个未知转录因子结合位点(TFBS)的候选调节因子,我们预测其TFBS。使用Yu等人的方法(18)和拟南芥转录因子的TFBS数据库(方法),我们能够预测ZmmTERF-1(Zm00001d031533,L7)、ZmARF1-2(Zm00001d003601,L8)、ZmWRKY39(Zm00001d013307,L8)和ZmMYB117(Zm00001d032194,L10)的TFBS(图3A中以红色表示)。预测的TFBS显示在附录SI,图S5中。对于图3A中以橙色表示的TF,我们无法预测其TFBS,因为玉米和拟南芥之间的DNA结合域差异过大或没有拟南芥的数据可用。
第三,对于每个具有已知或预测TFBS的TF,我们检查其TFBS在每个候选靶基因的启动子区域中的存在情况。这一过程进一步简化了图3A中的网络,变为图3B。
接下来,我们使用电泳迁移率变化分析(EMSA)和原生质体瞬时分析(PTA)来验证我们对候选调节因子及其假设靶基因的预测。我们的EMSA实验为ZmARF1-2与ZmWRKY39启动子的结合、ZmWRKY39与ZmMYB117启动子的结合以及ZmMYB117与ZmSHR1启动子的结合提供了证据(图3B和D-F),但未显示ZmmTERF-1与ZmARF1-2启动子的结合(图3C)。我们的PTA实验表明,ZmARF1-2的过表达上调了ZmWRKY39、ZmMYB117和ZmSHR1的表达;ZmWRKY39的过表达上调了ZmMYB117和ZmSHR1的表达;ZmMYB117的过表达上调了ZmSHR1的表达(图3B)。因此,我们的EMSA和PTA实验都支持图3B中红色箭头指示的上游调节级联;即,ZmARF1-2是ZmWRKY39的直接上游调节因子,而ZmWRKY39则是ZmMYB117的直接上游调节因子,然后ZmMYB117作为ZmSHR1的直接上游调节因子。
鉴定C4酶基因的调节因子
为展示我们方法的广泛实用性,我们分析了王等人的3D数据(6),这些数据是来自15和11段第三发育叶的玉米(C4)和水稻(C3)的两个系列转录组(数据集S3)。注意这里我们考虑的是空间点而不是时间点。由于所有关键的C4酶基因在束鞘或叶肉(M)细胞中优先表达,我们旨在鉴定关键C4酶基因的上游调节因子,每个基因在玉米叶中都有强烈的细胞类型表达偏好(19, 20)。按照图1A中的方法,我们首先确定玉米(标记为Zm+)和水稻(标记为Os+)中任何TF-基因对的正相关和无共表达的截止值,分别为PCC≥0.93(P < 0.05)和0.5≥ PCC >−0.5。其次,我们构建一个具有Zm+OS0共表达关系的玉米特异GCN。第三,我们根据第二步构建的GCN建立一个空间有序的GCN,并识别与那些关键C4酶基因共表达的TF基因(表1)。最后,通过EMSA验证候选的TF-靶标。
对于每个转录因子-候选靶基因对,我们确定转录因子结合位点(TFBS)是否可以在候选靶基因的启动子区域中找到。使用Yu等人的方法(18)和拟南芥TFBS数据库,我们预测了12个转录因子-靶基因对(表1和附录SI,表S4)。我们发现了九个可能调节BS偏好的C4酶基因的转录因子,包括四个转录因子(ZmGATA12、ZmbHLH43、ZmERF和ZmNAC)针对ZmNADP-ME(NADP-苹果酸酶,Zm00001d000316),四个转录因子(ZmMYB48、ZmMYB88、ZmMYB56和ZmbHLH118)针对ZmPCK(磷酸烯醇丙酮酸羧化酶,Zm00001d028471),以及一个转录因子(ZmMYBr17)针对ZmRBCS2(表1)。我们发现的ZmNADP-ME的四个转录因子与Borba等人(21)鉴定的两个转录因子ZmbHLH128(Zm00001d054038)和ZmbHLH129(Zm00001d014995)不同,这两个转录因子在BS或M细胞中没有强烈的偏好表达(20)。对于M偏好的C4酶基因,我们发现两个转录因子(ZmABI33和ZmRAV)可能上调ZmCA(碳酸酐酶,Zm00001d044099)的表达,且ZmRAV也可能上调ZmPEPC(磷酸烯醇丙酮酸羧化酶,Zm00001d046170)的表达(表1)。
对于EMSA验证,七种重组转录因子蛋白(ZmGATA12、ZmbHLH43、ZmMYB88、ZmMYB56、ZmMYB48、ZmbHLH118和ZmMYBr17)已成功表达、纯化,并用于评估三个候选靶基因(ZmNADP-ME、ZmPCK和ZmRBCS2)的直接转录因子-靶基因相互作用(表1)。我们的EMSA实验验证了ZmGATA12和ZmbHLH43与ZmNADP-ME的结合(附录SI,图S6 A和B)、ZmMYB88、ZmMYB56、ZmMYB48和ZmbHLH118与ZmPCK的结合(附录SI,图S6 C–F)以及ZmMYBr17与ZmRBCS2的结合(附录SI,图S6G)。ZmMYB88、ZmMYB56和ZmMYB48与ZmPCK相同TFBS的结合支持了具有相似DNA结合域的转录因子结合相似DNA序列基序的观点(22)。
讨论
本研究的主要贡献是一种比较不同条件(或组织/器官或物种)和时间点(或空间点)下获得的3D转录组的方法。我们的方法具有以下优势。首先,它可以轻松识别每个条件下共表达的基因对,然后找出哪些共表达关系在条件之间得以保留。其次,不需要在条件之间标准化RPKM值。如果条件之间的发育动态非常不同,标准化可能很困难。第三,不需要在两个条件之间对齐时间或空间点。因此,研究的样本点数可以在条件之间有所不同。我们将该方法应用于从15个和11个发育中的玉米和水稻叶片段获得的两组转录组的例子提供了这一点的证明。第四,我们的方法可以减少批次效应,因为在构建GCNs之前,共表达是在每组转录组内定义的。出于这个原因,这种方法可以应用于对来自不同实验室的异质转录组数据集进行元分析。第五,最重要的是,我们的方法提供了TO-GCNs,可以揭示受环境条件影响的发育转变背后的基因表达的时间动态。此外,TO-GCNs提供了一种推断任何感兴趣基因的候选上游调节因子的便捷方式,如果该基因至少在一个TO-GCN中存在。
我们测试了当用一个不同于ZmARF1-2的转录因子基因作为初始节点来构建TO-GCN时的级别顺序稳定性。我们随机选择了原始TO-GCN第一级的10个不同转录因子基因,并逐一测试它们。我们计算了每个测试转录因子基因与原始基因相比的级别差异。结果显示,平均约12.5%的原始TO-GCN中的转录因子被分配到不同的级别(附录SI,表S5)。然而,每个新TO-GCN的整体级别变化的平均值和SD非常小(附录SI,表S5),表明新的有序TO-GCN与原始的非常相似。
除了提供一种方法外,我们的研究有助于更好地理解玉米早期叶片发育中的光独立过程。超过1,200个转录因子基因被分配到光独立的TO-GCN中,提供了光独立基因调节关系的全局图景。通常,预测上游调节因子比预测下游靶基因要困难得多。在本研究中,我们展示了我们的方法可以成功地识别从ZmARF1-2(L8)到ZmSHR1(L11)的关键克兰茨解剖结构调节因子的上游调节级联,这与生长素在克兰茨解剖结构发育中发挥重要作用的事实相符(17)。此外,我们比较了ZmARF1-2、ZmWRKY39、ZmMYB117和ZmSHR1在玉米叶片和苞叶发育中的表达谱。这些比较可能揭示了这些转录因子基因在我们研究的C4和C3植物中是如何进化的,因为在玉米中,叶片和苞叶分别表现出C4和C3光合作用。Wang等人(3)获得了叶片和苞(P1、P2、P3/4、P5、I和E;附录SI,图S7)六个发育阶段的转录组。我们发现ZmARF1-2、ZmWRKY39和ZmMYB117具有相似的表达谱,在早期胚胎阶段(P1、P2和P3/4在附录SI,图S7)的叶片中表达量远高于苞叶,这是克兰茨解剖结构发育的阶段。这一观察表明,这三个TF基因的调节在C4叶的进化过程中发生了变化。ZmARF1-2、ZmWRKY39和ZmMYB117与ZmSHR1在叶片中的表达谱的相似性以及在苞叶中的不同(附录SI,图S7)表明,这三个TF可能在叶片(C4)中而不是在苞叶(C3)中调节ZmSHR1。这一观察与我们的提议一致,即ZmARF1-2、ZmWRKY39和ZmMYB117是ZmSHR1的上游调节因子。ZmSHR1的一些直接和间接靶基因曾在拟南芥根血管发育的研究中被报道(23)。我们发现ZmMGP/NUC(MAGPIE/NUTCRACKER;Zm00001d009030)和ZmSCR1,ZmSHR1的两个直接靶标分别位于L11和L12,以及ZmSCL3(SCR-LIKE 3;Zm00001d011881),两个ZmSNE的同源基因(SNEEZY;Zm00001d028159和Zm00001d048185)以及两个ZmRLK的同源基因(RECEPTOR-LIKE KINASE;Zm00001d005298和Zm00001d046626),以及ZmSHR1的五个间接靶标位于L11、L12或L13,表明ZmSHR1在双子叶根的调节途径在单子叶叶中被广泛保守。除了时间序列数据外,我们还将我们的方法应用于比较玉米和水稻之间的叶发育系列的另一个3D基因表达数据集,从而识别了一个“空间有序”的GCN以及新的调节因子-靶关系,进一步支持了我们方法的价值。
总之,我们展示了一种用于分析3D数据集的强大方法的实用性。它可以应用于对比广泛情境中的基因共表达谱。考虑到实验设计日益复杂的基因表达数据的迅速涌入,我们的方法为挖掘这些表达数据以获得生物学洞见提供了手段。通过将我们的方法应用于玉米叶在LD和TD条件下的时间表达数据以及玉米和水稻的叶片发育转录组,推断出TO-GCNs,提供了丰富的调节相互作用预测。结合实验验证,我们进一步揭示了C4光合作用中叶脉发育的关键调节级联。这些发现不仅突显了我们方法预测的调节相互作用质量,还提供了关于C3-C4进化转变的调节基础的必要信息,为未来用遗传工程改造C3作物具有C4光合作用的能力铺平了道路(24, 25)。
方法
RNA测序和读取处理
购买了当地供应商提供的Zea mays cv. White Crystal种子,这是一种糯玉米品种。为了使种子发芽,将种子浸泡在蒸馏水中,在晚上6:00进行200 rpm摇晃10分钟,然后在暗室中的培养皿上湿滤纸上进行发芽,温度为30°C,湿度为60%。在暗室中的昏暗绿光下每6小时收集一次胚芽,一小时内去除胚芽鞘,胚胎叶冷冻于液氮中并储存在-80°C。
使用TRIzol试剂(Invitrogen)提取总RNA。为了去除DNA污染的痕迹,向每10μg RNA中加入1μL TURBO DNase(Ambion),在37°C下孵育30分钟,然后进行苯酚:氯仿提取。使用BioAnalyzer RNA 6000 Nano Kit(Agilent)测定RNA样品的数量和质量。使用TruSeq RNA Library Prep Kit v2(Illumina)构建RNA测序文库。接头连接的反应选择两个大小范围(约300和400 bp)。使用AMPure XP珠子(Beckman Agencourt)清理纯化后的文库,然后使用Qubit HS DNA Kit和BioAnalyzer HS DNA Kit(Agilent)进行检测,并使用KAPA Library Quantification Kit Illumina Platforms(Kapa Biosystems)进行摩尔浓度标准化。在Academia Sinica的NGS High Throughput Genomics Core Facility使用Illumina HiSeq 2000进行配对末端2×101-nt测序。原始读取数据存放在Short Read Archive中,https://www.ncbi.nlm.nih.gov/sra(登录号SRP140487)。读取处理过程与Liu等人(4)相同。处理后的读取使用TopHat(26)(v2.0.10)和其嵌入式比对器Bowtie2(27)(v2.1.0)映射到玉米基因组(B73 RefGen_v4)。使用Cufflinks(28)(v2.1.1)估计每个基因的表达水平(RPKM)。为了比较一组转录组中所选基因在时间点上的RPKM,我们应用了上四分位数归一化程序(29)。
构建GCN
我们的比较转录组学方法旨在分析可能在两个或更多条件下具有不同时间点数量的时间序列转录组。该方法包括三个步骤:确定共表达截止值、构建GCN、确定转录因子基因表达的时间顺序(图1)。在这项研究中,输入数据是来自LD和TD条件下玉米胚胎叶的两个时间序列转录组,以及Dataset S4中的玉米转录因子基因列表[从Lin等人(30)更新]。首先,分别在LD和TD下计算所有转录因子基因对的皮尔森相关系数值,并用于确定正共表达(标记为LD+或TD+)、负共表达(标记为LD−或TD−)和无共表达(标记为LD0或TD0)的截止值(结果部分的示例)。其次,使用这三种关系类型,确定所有类型的GCN(附录SI,表S2)。在这项研究中,我们专注于具有1,207个节点的主要GCN,带有LD+TD+共表达关系;其他LD+TD+ GCN的节点少于10个。这个GCN是光独立的,因为这个GCN中的所有共表达关系都不受光的存在或缺失的影响。第三,通过
从一个选定的节点开始的广度优先搜索算法(9)分配每个GCN中的转录因子基因的时间顺序。BFS是一种搜索网络图的算法。它从一个初始种子开始,并搜索其所有邻居(有连接边的节点)以形成一组节点(第1级)。然后,过程从第1级的所有节点开始,搜索它们的邻居(排除第1级的节点)以形成另一组节点(第2级),依此类推,直到网络中的所有节点都被分配。该方法的计算程序可在GitHub - petitmingchang/TO-GCN: Pipeline of time-ordered gene coexpression network (TO-GCN) construction from three-dimensional (gene expression, condition, and time) data(31)获取。
如前所述,对于光独立的GCN,选择ZmARF2-1作为初始节点。根据BFS算法,ZmARF2-1及与其共表达的所有节点被分配到第1级(标记为L1)。然后,与L1中任何节点共表达的所有节点被分配到L2,与L2中任何节点共表达的所有节点被分配到L3,依此类推,直到GCN中的所有节点都被分配。
每个TO-GCN级别中共表达基因集和过表达功能
对于TO-GCN中每个级别的转录因子基因,可以识别具有相同共表达关系的共表达基因集,以将基因添加到TO-GCN中。由于一个基因可能与多个级别的转录因子共表达,因此两个相邻基因集将具有一些重叠的基因(图1B)。
对于与TO-GCN中某一级别相对应的每一组基因,使用这项研究中表达的所有基因作为背景集进行功能富集分析。使用Fisher精确测试,误差发现率(FDR)<0.05(32),并使用MapMan的功能注释(Home - MapMan)(图1B)。
预测玉米转录因子基因及其靶基因的结合位点
为预测玉米转录因子基因的结合位点,我们收集了已知的拟南芥转录因子基因的TFBS(位置权重矩阵;PWMs)信息,包括CIS-BP(22)、JASPAR(33)、Plant Cistrome(34)、Franco-Zorrilla等人(35)和Sullivan等人(36)的数据库和文献。如果一个转录因子有来自不同来源的多个PWM,我们选取信息含量最高的PWM。由于具有相似DNA结合域(DBDs)的转录因子具有相似的DNA序列偏好,对于一个玉米转录因子,我们使用其DBD来识别具有已知TF-TFBS对的拟南芥DBDs的同源物。使用Yu等人的方法2(18),我们发现了在LD和TD下与玉米转录因子共表达且PCC >0.8的基因,并对这些基因进行了基因集富集分析。对于在基因集中富集的基因的启动子,我们鉴定了过表达的基序(PWMs),并在四个参考物种(短柄草、水稻、高粱和刚毛狗尾草)中测试了它们的保守性,P值 < 10−5。然后通过的PWMs被视为假定的转录因子结合基序(TFBMs,PWMs)。对于一个转录因子识别的假定TFBMs中,最类似于已知拟南芥转录因子(与玉米转录因子DBD序列相似度>70%)TFBM的TFBM被视为该转录因子的主要TFBM。然后我们将玉米转录因子的主要TFBMs应用于预测TF靶基因,通过检查启动子区域中TFBSs(由TFBMs映射定位)的保守性来完成,如Yu等人(18)所描述。基因的启动子区域定义为基因转录起始位点上游的1-kb序列。例如,在鉴定C4酶基因的调节因子中,我们发现ZmbHLH43的DBD与拟南芥PIF3(AT1G09530)的DBD有79%的同一性,后者在CIS-BP中有已知的DNA结合基序(ID M2863 1.02)。ZmNADP-ME的启动子包含了在四个参考物种中三个NADP-ME启动子的保守测试通过的DNA序列。
载体构建
用于EMSA的,预测的TF的全长cDNA被克隆进pet42a载体,使用设计的引物对(附录SI,表S6和S7)。该质粒用于转化大肠杆菌Rosetta(DE3)。对于原生质体转染,将GFP和预测的玉米TFs的全长cDNA在玉米泛素1启动子的控制下克隆进pBI221载体,使用设计的引物对(附录SI,表S6)。使用Maxi Plasmid Kit(Qiagen)纯化的质粒DNA用于转染。
EMSA验证
如Yu等人(18)描述的方法进行,略有修改。与1-(约50 ng)、2-或4-倍的GST或从大肠杆菌Rosetta(DE3)中表达和纯化的重组TF蛋白在22°C下孵育20分钟的生物素标记探针(附录SI,表S8和S9)进行孵育。竞争实验使用1-(10 ng)、5-或10-倍的未标记探针作为竞争物进行。EMSA混合物通过3.75%聚丙烯酰胺天然凝胶分离,并通过半干转移池(Bio-Rad)转移到Hybond N+膜(GE)。通过链霉亲和素-HRP偶联物(Life Technologies)与ECL Plus(GE)底物检测生物素标记的探针和TF-探针复合物。化学发光信号通过BioSpectrum成像系统(UVP)可视化。
原生质体瞬时分析
如Chang等人(20)描述的方法进行,略有修改。从幼嫩的黄化玉米幼苗的叶子中分离出叶肉原生质体。细胞浓度调整至每毫升5×10^5,200 μL原生质体与质粒DNA(5 μg pBI221-GFP与10 μg pBI221或10 μg pBI221-TF)混合。加入等体积的PEG溶液(0.6 M甘露醇,0.1 M氯化钙和40% PEG 4000),轻轻翻转混合物。在室温下孵育20分钟后,通过离心(150×g,2分钟)收集原生质体,并在500 μL孵育液中重新悬浮(0.6 M甘露醇,4 mM氯化钾和4 mM Mes,pH 5.7)。转染的原生质体转移到Falcon培养板中,在26°C的黑暗中孵育6小时(37)。通过离心(150×g,2分钟)收集原生质体,用于总RNA提取。
RNA提取和qRT-PCR
如Chang等人(20)描述的方法进行,提取原生质体中的总RNA,并使用NanoDrop(Thermo Scientific)和甲醛凝胶电泳检查其质量。使用SuperScript III First-Strand Synthesis SuperMix Kit(Invitrogen)从0.5 μg RNA合成一链cDNA。使用0.01 μg cDNA在LightCycler 480 Instrument System(Roche)上进行qRT-PCR,使用KAPA SYBR FAST qPCR Master Mix进行,初始变性步骤在95°C下进行5分钟,然后进行55个周期,每个周期包括95°C下10秒,60°C下20秒和72°C下5秒。qRT-PCR用于的引物列在附录SI,表S10中。首先,计算肌动蛋白基因和目标基因之间的循环阈值(Ct)值的差异,计为deltaCtcontrol(pBI221-GFP与pBI221处理)或deltaCtTF(pBI221-GFP与pBI221-TF处理)。然后,计算这些值之间的差异,表示为2deltaCT(TF - control)。